
IMFINZI, 50 mg-ml solution à diluer pour perfusion, boîte de 3 flacons de 10 ml
Retiré du marché le : 26/05/2021
Dernière révision : 13/03/2020
Taux de TVA : 0%
Laboratoire exploitant : ASTRAZENECA
Source :

IMFINZI, en association à l'étoposide et aux sels de platine (carboplatine ou cisplatine), est indiqué dans le traitement de première intention des patients atteints d'un cancer bronchique à petites cellules à un stade étendu (CBPC-SE).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom de marque et le numéro de lot du produit administré doivent être clairement enregistrés.
Pneumopathie immuno-médiée
Une pneumopathie immuno-médiée ou pneumopathie interstitielle diffuse, définie comme nécessitant l'utilisation de corticoïdes systémiques et sans autre étiologie évidente, est survenue chez des patients recevant IMFINZI.
Une pneumopathie radique est fréquemment observée chez les patients recevant une radiothérapie des poumons et la présentation clinique de la pneumopathie immuno-médiée est très similaire à celle de la pneumopathie radique.
Les patients doivent être surveillés afin de déceler tous signes et symptômes de pneumopathie (immuno-médiée ou radique). Les patients avec une suspicion de pneumopathie doivent être évalués par imagerie radiographique et traités conformément aux recommandations de la rubrique Posologie et mode d'administration.
Hépatite immuno-médiée
Des cas d'hépatite immuno-médiée, définie comme nécessitant l'utilisation de corticoïdes systémiques et sans autre étiologie évidente, ont été décrits chez des patients recevant IMFINZI (voir rubrique Effets indésirables). Les patients doivent être surveillés par des tests de la fonction hépatique pour déceler d'éventuelles anomalies, avant et périodiquement pendant le traitement par IMFINZI, et tel qu'indiqué en fonction de l'évaluation clinique. L'hépatite immuno-médiée doit être traitée conformément aux recommandations de la rubrique Posologie et mode d'administration.
Colite immuno-médiée
Des cas de colite ou de diarrhée immuno-médiée, définie comme nécessitant l'utilisation de corticoïdes systémiques et sans autre étiologie évidente, ont été décrits chez des patients recevant IMFINZI (voir rubrique Effets indésirables). Les patients doivent être surveillés afin de déceler tous signes et symptômes de colite ou de diarrhée et traités conformément aux recommandations de la rubrique Posologie et mode d'administration.
Endocrinopathies immuno-médiées
Hypothyroïdie, hyperthyroïdie et thyroïdite immuno-médiées
Des cas de dysthyroïdies immuno-médiées (hypothyroïdie, hyperthyroïdie et thyroïdite) sont survenus chez des patients recevant IMFINZI, et une hypothyroïdie peut suivre une hyperthyroïdie (voir rubrique Effets indésirables). Les patients doivent être surveillés par des tests de la fonction thyroïdienne pour déceler d'éventuelles anomalies, avant et périodiquement au cours du traitement par IMFINZI et tel qu'indiqué en fonction de l'évaluation clinique. Ces dysthyroïdies immuno-médiées (hyperthyroïdie, hypothyroïdie et thyroïdite) doivent être traitées conformément aux recommandations de la rubrique Posologie et mode d'administration.
Insuffisance surrénalienne immuno-médiée
Des cas d'insuffisance surrénalienne immuno-médiée ont été décrits chez des patients recevant IMFINZI (voir rubrique Effets indésirables). Les patients doivent être surveillés afin de déceler tous signes et symptômes cliniques d'insuffisance surrénalienne. En cas d'insuffisance surrénalienne symptomatique, les patients doivent être traités conformément aux recommandations de la rubrique Posologie et mode d'administration.
Diabète sucré de type 1 immuno-médié
Des cas de diabète sucré de type 1 immuno-médié ont été décrits chez des patients recevant IMFINZI (voir rubrique Effets indésirables). Les patients doivent être surveillés afin de déceler tous signes et symptômes cliniques de diabète sucré de type 1. En cas de diabète sucré de type 1 symptomatique, les patients doivent être traités conformément aux recommandations de la rubrique Posologie et mode d'administration.
Hypophysite / hypopituitarisme immuno-médié(e)
Des cas d'hypophysite ou d'hypopituitarisme immuno-médié ont été décrits chez des patients recevant IMFINZI (voir rubrique Effets indésirables). Les patients doivent être surveillés afin de déceler tous signes et symptômes cliniques d'hypophysite ou d'hypopituitarisme. En cas d'hypophysite ou d'hypopituitarisme symptomatique, les patients doivent être traités conformément aux recommandations de la rubrique Posologie et mode d'administration.
Néphrite immuno-médiée
Des cas de néphrite immuno-médiée, définie comme nécessitant l'utilisation de corticoïdes systémiques et sans autre étiologie évidente, ont été décrits chez les patients recevant IMFINZI (voir rubrique Effets indésirables). Les patients doivent être surveillés par une évaluation de la fonction rénale pour déceler d'éventuelles anomalies avant et périodiquement pendant le traitement par IMFINZI et traités conformément aux recommandations de la rubrique Posologie et mode d'administration.
Éruption cutanée immuno-médiée
Des cas d'éruptions cutanées ou de dermatites immuno-médiées (incluant des pemphigoïdes), définies comme nécessitant l'utilisation de corticoïdes systémiques et sans autre étiologie évidente, ont été décrits chez des patients recevant IMFINZI (voir rubrique Effets indésirables). Des cas de Syndrome de Stevens-Johnson ou de nécrolyse épidermique toxique ont été rapportés chez des patients traités par des inhibiteurs PD-1. Les patients doivent être surveillés afin de déceler tous signes et symptômes d'éruption cutanée ou de dermatite et être traités conformément aux recommandations de la rubrique Posologie et mode d'administration.
Autres effets indésirables immuno-médiés
Compte tenu du mécanisme d'action d'IMFINZI, d'autres effets indésirables immuno-médiés éventuels peuvent survenir. Les effets indésirables immuno-médiés suivants ont été rapportés chez moins de 1% des patients traités par IMFINZI en monothérapie dans les essais cliniques (n = 3 006) : myasthénie grave, myocardite, myosite, polymyosite. Les patients doivent être surveillés afin de déceler tous signes et symptômes de myocardite, myosite ou polymyosite et être traités conformément aux recommandations de la rubrique Posologie et mode d'administration. Des cas de pancréatite ont été rapportés chez des patients ayant reçu IMFINZI dans le cadre des essais cliniques. Les patients doivent être surveillés afin de déceler tous signes et symptômes et être traités, pour les autres effets indésirables immuno-médiés, conformément aux recommandations en rubrique Posologie et mode d'administration.
Réactions liées à la perfusion
Les patients doivent être surveillés afin de déceler tous signes et symptômes de réactions liées à la perfusion. Des cas de réactions sévères liées à la perfusion ont été rapportés chez des patients recevant IMFINZI (voir rubrique Effets indésirables). Des réactions liées à la perfusion doivent être prises en charge selon les recommandations en rubrique Posologie et mode d'administration.
Patients exclus des essais cliniques
Les patients présentant les conditions suivantes à l'inclusion ont été exclus des essais cliniques : statut de performance ECOG ≥ 2 ; une maladie auto-immune active ou précédemment documentée au cours des 2 ans ayant précédé le début de l'étude ; un antécédent d'immunodéficience ; un antécédent d'effets indésirables immuno-médiés sévères ; pathologies nécessitant une immunosuppression systémique, à l'exception de doses physiologiques de corticostéroïdes systémiques (≤ 10 mg/jour de prednisone ou équivalent) ; tuberculose active ou hépatite B ou C ou infection par le VIH ou patients devant recevoir un vaccin vivant atténué dans les 30 jours avant ou après le début d'IMFINZI. En l'absence de données, le durvalumab doit être utilisé avec prudence chez ces populations après une évaluation attentive du bénéfice/risque potentiel sur une base individuelle.
Résumé du profil de tolérance
La tolérance d'IMFINZI en monothérapie repose sur les données poolées obtenues chez 3 006 patients présentant de nombreux types de tumeurs. IMFINZI a été administré à une dose de 10 mg/kg toutes les 2 semaines ou de 20 mg/kg toutes les 4 semaines.
Les effets indésirables les plus fréquents (>10 %) étaient : toux/toux productive (21,5 %), diarrhée (16,3 %), éruption cutanée (16,0 %), fièvre (13,8 %), infections des voies respiratoires supérieures (13,5 %), douleur abdominale (12,7 %), prurit (10,8 %) et hypothyroïdie (10,1 %).
La
tolérance d'IMFINZI administré en association avec une chimiothérapie
repose sur les données obtenues chez 265 patients présentant un CBPC.
IMFINZI a été administré à une dose de 1 500 mg toutes les 3 ou 4
semaines. Les effets indésirables les plus fréquents (> 20 %)
étaient : neutropénie (48,7 %), anémie (38,5 %), nausées (33,6 %),
fatigue (32,1 %), alopécie (31,3 %), thrombocytopénie (21,1 %) et
leucopénie (20,0 %).
Liste tabulée des effets indésirables
Le tableau 3 liste l'incidence des effets indésirables de l'ensemble des données de tolérance en monothérapie et chez les patients traités par IMFINZI en association avec une chimiothérapie dans l'étude CASPIAN. Les effets indésirables sont classés selon la classe de systèmes d'organes de MedDRA. Au sein des classes de systèmes d'organes, les effets indésirables sont présentés suivant un ordre de fréquence décroissant. La catégorie de fréquence correspondante pour chaque effet indésirable est définie comme : très fréquent (≥1/10), fréquent (≥1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupement de fréquence, les effets indésirables sont présentés par ordre de gravité décroissant.
Tableau 3. Effets indésirables chez les patients traités par IMFINZI en monothérapie et IMFINZI en association avec une chimiothérapie
| | IMFINZI en monothérapie | IMFINZI en association avec une chimiothérapie | ||||
| | Tous grades (%) | Grade 3-4 (%) | Tous grades (%) | Grade 3-4 (%) | ||
| Infections et infestations | ||||||
| Infections des voies respiratoires supérieuresa | Très fréquent | 13,5 | 0,2 | Fréquent | 9,1 | 0,4 |
| Pneumonieb,c | Fréquent | 8,9 | 3,5 | Fréquent | 5,7 | 1,9 |
| Candidose buccale | Fréquent | 2,1 | 0 | Peu fréquent | 0,8 | 0 |
| Infections dentaires et des tissus mous buccauxd | Fréquent | 1,7 | <0,1 | Fréquent | 1,1 | 0 |
| Grippe | Fréquent | 1,6 | <0,1 | Peu fréquent | 0,4 | 0 |
| Affections hématologiques et du système lymphatique | ||||||
| Neutropéniee | | | | Très fréquent | 48,7 | 29,1 |
| Anémie | | | | Très fréquent | 38,5 | 9,1 |
| Thrombocytopénief | | | | Très fréquent | 21,1 | 6,8 |
| Leucopénieg | | | | Très fréquent | 20,0 | 7,9 |
| Neutropénie fébrile | | | | Fréquent | 6,4 | 5,3 |
| Pancytopénie | | | | Fréquent | 3,0 | 1,5 |
| Affections endocriniennes | ||||||
| Hypothyroïdieh | Très fréquent | 10,1 | 0,2 | Fréquent | 9,4 | 0 |
| Hyperthyroïdiei | Fréquent | 4,6 | 0 | Fréquent | 9,8 | 0 |
| Thyroïditej | Peu fréquent | 0,8 | <0,1 | Fréquent | 1,5 | 0 |
| Insuffisance surrénalienne | Peu fréquent | 0,6 | <0,1 | Fréquent | 1,1 | 0 |
| Diabète sucré de type 1 | Rare | <0,1 | <0,1 | Peu fréquent | 0,8 | 0,8 |
| Hypopituitarisme/ hypophysite | Rare | <0,1 | <0,1 | Non déterminé | 0 | 0 |
| Diabète insipide | Rare | <0,1 | <0,1 | Non déterminé | 0 | 0 |
| Affections du système nerveux | ||||||
| Myasthénie grave | Rarek | <0,1 | <0,1 | | | |
| Troubles du métabolisme et de la nutrition | ||||||
| Baisse de l'appétit | | | | Très fréquent | 18,1 | 0,8 |
| Affections cardiaques | | | | |||
| Myocardite | Rare | <0,1 | <0,1 | Non déterminé | 0 | 0 |
| Affections respiratoires, thoraciques et médiastinales | ||||||
| Toux / toux productivel | Très fréquent | 21,5 | 0,4 | Très fréquent | 14,7 | 0,8 |
| Pneumopathie inflammatoireb | Fréquent | 3,8 | 0,9 | Fréquent | 2,6 | 0,8 |
| Dysphonie | Fréquent | 3,1 | <0,1 | Peu fréquent | 0,8 | 0 |
| Pneumopathie interstitielle diffuse | Peu fréquent | 0,6 | 0,1 | Peu fréquent | 0,8 | 0 |
| Affections gastro-intestinales | ||||||
| Diarrhée | Très fréquent | 16,3 | 0,6 | Fréquent | 9,8 | 1,1 |
| Douleur abdominalem | Très fréquent | 12,7 | 1,8 | Fréquent | 8,7 | 0,4 |
| Coliten | Peu fréquent | 0,9 | 0,3 | Peu fréquent | 0,8 | 0 |
| Nausées | | | | Très fréquent | 33,6 | 0,4 |
| Constipation | | | | Très fréquent | 16,6 | 0,8 |
| Vomissements | | | | Très fréquent | 14,7 | 0 |
| Stomatiteo | | | | Fréquent | 6,0 | 0,4 |
| Affections hépatobiliaires | ||||||
| Aspartate aminotransférase augmentée ou Alanine aminotransférase augmentéep | Fréquent | 8,1 | 2,3 | Fréquent | 8,7 | 1,9 |
| Hépatitec,q | Peu fréquent | 0,8 | 0,4 | Fréquent | 1,9 | 1,1 |
| Affections de la peau et du tissu sous-cutané | ||||||
| Éruption cutanéer | Très fréquent | 16,0 | 0,6 | Fréquent | 9,4 | 0 |
| Prurits | Très fréquent | 10,8 | <0,1 | Fréquent | 7,5 | 0 |
| Sueurs nocturnes | Fréquent | 1,6 | <0,1 | Peu fréquent | 0,4 | 0 |
| Dermatitet | Peu fréquent | 0,7 | <0,1 | Fréquent | 1,5 | 0 |
| Alopécie | | | | Très fréquent | 31,3 | 1,1 |
| Pemphigoïdeu | Peu fréquent | 0,2 | 0 | | | |
| Affections musculo-squelettiques et du tissu conjonctif | ||||||
| Myalgie | Fréquent | 5,9 | <0,1 | Fréquent | 3,4 | 0 |
| Myosite | Peu fréquent | 0,2 | <0,1 | Non déterminé | 0 | 0 |
| Polymyositec | Rareg | <0,1 | <0,1 | Non déterminé | 0 | 0 |
| Affections du rein et des voies urinaires | ||||||
| Créatininémie augmentée | Fréquent | 3,5 | <0,1 | Fréquent | 1,9 | 0 |
| Dysurie | Fréquent | 1,3 | 0 | Fréquent | 1,9 | 0 |
| Néphritev | Peu fréquent | 0,3 | <0,1 | Non déterminé | 0 | 0 |
| Troubles généraux et anomalies au site d'administration | ||||||
| Fièvre | Très fréquent | 13,8 | 0,3 | Fréquent | 8,3 | 0 |
| Œdème périphériquew | Fréquent | 9,7 | 0,3 | Fréquent | 6,4 | 0,8 |
| Fatiguex | | | | Très fréquent | 32,1 | 3,4 |
| Lésions, intoxication et complications liées aux procédures | ||||||
| Réaction liée à la | Fréquent | 1,6 | 0,2 | Fréquent | 1,9 | 0,4 |
| perfusiony | | | | | | |
a inclut laryngite, nasopharyngite, abcès périamygdalien, pharyngite, rhinite, sinusite, angine, trachéobronchite et infection des
voies aériennes supérieures.
b inclut infection pulmonaire, pneumonie à pneumocystis jirovecii, pneumonie, pneumonie à adénovirus, pneumonie bactérienne, pneumonie à cytomégalovirus, pneumonie à haemophilus, pneumonie à pneumocoques, pneumonie à streptocoques, pneumonie à candida et pneumonie à legionella.
c y compris issue fatale.
d inclut gingivite, infection buccale, parodontite, pulpite dentaire, abcès dentaire et infection dentaire.
e inclut neutropénie et baisse du taux de neutrophiles.
f inclut thrombocytopénie et baisse du taux de plaquettes.
g inclut leucopénie et baisse du taux de globules blancs.
h inclut hypothyroïdie et hypothyroïdie auto-immune.
i inclut hyperthyroïdie et maladie de Basedow.
j inclut thyroïdite auto-immune, thyroïdite et thyroïdite subaiguë.
k la fréquence est basée sur des événements non observés dans l'étude PACIFIC mais observés dans les essais cliniques d'AstraZeneca (n= 4067).
l inclut toux et toux productive.
m inclut douleur abdominale, douleur abdominale basse, douleur abdominale haute et douleur du flanc.
n inclut colite, entérite, entérocolite et proctite.
o inclut stomatite et inflammation des muqueuses.
p inclut alanine aminotransférase augmentée, aspartate aminotransférase augmentée, enzymes hépatiques augmentées et transaminases augmentées.
q inclut hépatite, hépatite auto-immune, hépatite toxique, lésion hépatocellulaire, hépatite aiguë, hépatotoxicité et hépatite immuno-médiée.
r inclut éruption cutanée érythémateuse, éruption cutanée généralisée, éruption cutanée maculeuse, éruption cutanée maculopapuleuse, éruption cutanée papuleuse, éruption cutanée prurigineuse, éruption cutanée pustuleuse, érythème, eczéma et éruption cutanée.
s inclut prurit généralisé et prurit.
t inclut dermatite bulleuse et pemphigus. La fréquence observée dans les essais terminés et en cours est peu fréquent.
u la fréquence est basée sur un événement de dermatite bulleuse observé dans l'étude PACIFIC. D'autres événements de pemphigoïde, de dermatite bulleuse et de pemphigus ont été observés dans les essais cliniques terminés et en cours, la fréquence rapportée est peu fréquente.
v inclut néphrite auto-immune, néphrite tubulo-interstitielle, néphrite, glomérulonéphrite et glomérulonéphrite extramembraneuse.
w inclut oedème périphérique et gonflement périphérique.
x inclut fatigue et asthénie.
y inclut réaction à la perfusion et urticaire débutant le jour du traitement ou 1 jour après.
Description des effets indésirables sélectionnés
IMFINZI est le plus souvent associé à des effets indésirables immuno-médiés. La plupart d'entre eux, y compris les effets sévères, se sont résolus après l'initiation d'une thérapie médicale appropriée ou de l'arrêt d'IMFINZI. Les données correspondant aux effets indésirables immuno-médiés suivants reflètent la base de données de tolérance combinées de 3 006 patients dont l'étude PACIFIC et des études supplémentaires chez des patients atteints de tumeurs solides variées, dans des indications pour lesquelles le durvalumab n'est pas autorisé. Dans toutes les études, IMFINZI a été administré à une dose de 10 mg/kg toutes les 2 semaines, 20 mg/kg toutes les 4 semaines ou 1 500 mg toutes les 3 ou 4 semaines. Des détails sur les effets indésirables significatifs d'IMFINZI administré en association avec une chimiothérapie sont présentés lorsque des différences cliniquement pertinentes étaient notées comparativement à IMFINZI en monothérapie. Les recommandations de traitement de ces effets indésirables sont décrites dans la rubrique Mises en garde et précautions d'emploi.
Pneumopathie immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, (n = 3006 plusieurs types de tumeurs), une pneumopathie immuno-médiée est survenue chez 107 (3,6 %) patients, y compris de grade 3 chez 23 (0,8 %) patients, de grade 4 chez 2 (< 0,1 %) patients, et de grade 5 chez 6 (0,2 %) patients. Le temps médian de survenue a été de 57 jours (de 2 à 785 jours). Soixante-quatre patients sur 107 ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour), 2 patients ont également reçu de l'infliximab et 1 patient a également reçu de la ciclosporine. IMFINZI a été interrompu chez 38 patients. La résolution est survenue chez 58 patients.
Une pneumopathie immuno-médiée est survenue plus fréquemment chez les patients de l'étude PACIFIC ayant achevé le traitement par chimioradiothérapie concomitante dans les 1 à 42 jours précédant le début de l'étude (10,7 %), que chez les autres patients de la base de données combinées de tolérance (2,2 %).
Hépatite immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une hépatite immuno- médiée est survenue chez 36 (1,2 %) patients, y compris de grade 3 chez 19 (0,6 %) patients, de grade 4 chez 1 (< 0,1 %) patient et de grade 5 (fatal) chez 2 (< 0,1 %) patients. Le temps médian de survenue a été de 67 jours (de 7 à 333 jours). Vingt-cinq patients sur 36 ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Deux patients ont
également reçu un traitement par mycophénolate. IMFINZI a été interrompu chez 7 patients. La résolution est survenue chez 22 patients.
Colite immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une colite ou une diarrhée immuno-médiée est survenue chez 52 (1,7 %) patients, y compris de grade 3 chez 9 (0,3 %) patients, et de grade 4 chez 2 (< 0,1 %) patients. Le temps médian de survenue a été de 73 jours (de 1 à 394 jours). Trente-quatre patients sur 52 ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Un patient a également reçu un traitement par infliximab et un patient a également reçu du mycophénolate. IMFINZI a été interrompu chez 9 patients. La résolution est survenue chez 39 patients.
Endocrinopathies immuno-médiées Hypothyroïdie immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une hypothyroïdie immuno-médiée est survenue chez 222 (7,4 %) patients, y compris de grade 3 chez 4 (0,1 %) patients. Le temps médian de survenue a été de 85 jours (de 1 à 562 jours). Parmi les 222 patients, 218 ont reçu un traitement hormonal substitutif, 5 patients ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour) pour cause d'hypothyroïdie immuno-médiée suivi d'un traitement hormonal substitutif. Aucun patient n'a arrêté de prendre IMFINZI en raison d'une hypothyroïdie immuno-médiée.
Hyperthyroïdie immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une hyperthyroïdie immuno-médiée est survenue chez 43 (1,4 %) patients, et aucun cas de grade 3 ou 4 n'a été observé. Le temps médian de survenue a été de 43 jours (de 1 à 196 jours). 39 patients sur 43 ont reçu un traitement médical (thiamazole, carbimazole, propylthiouracile, perchlorate, inhibiteur calcique ou bêtabloquant), 11 patients ont reçu un traitement par corticoïdes systémiques et 4 de ces 11 patients ont reçu un traitement par corticoïdes systémiques à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Un patient a arrêté IMFINZI en raison d'une hyperthyroïdie. La résolution est survenue chez 35 patients. Dix-huit patients ont présenté une hypothyroïdie à la suite d'une hyperthyroïdie.
Thyroïdite immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une thyroïdite immuno-médiée est survenue chez 11 (0,4 %) patients, y compris de grade 3 chez 2 (< 0,1 %) patients. Le temps médian de survenue a été de 41 jours (de 14 à 106 jours). Sur les 11 patients, 9 ont reçu un traitement hormonal substitutif, 1 patient a reçu des corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour) suivi d'un traitement hormonal substitutif. Un patient a arrêté de prendre IMFINZI en raison d'une thyroïdite immuno-médiée. Deux patients ont présenté une hypothyroïdie après la thyroïdite.
Insuffisance surrénalienne immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une insuffisance surrénalienne immuno-médiée est survenue chez 12 (0,4 %) patients, y compris de grade 3 chez 2 (< 0,1 %) patients. Le temps médian de survenue a été de 145,5 jours (de 20 à 547 jours). Les 12 patients ont tous reçu un traitement par corticoïdes systémiques, 4 d'entre eux ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Aucun patient n'a arrêté de prendre IMFINZI en raison d'une insuffisance surrénalienne immuno-médiée. La résolution est survenue chez3 patients.
Diabète sucré de type 1 immuno-médié
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, un diabète sucré de type 1 immuno-médié est survenu chez 1 (< 0,1 %) patient, y compris de grade 3 chez 1 (< 0,1 %) patients. Le temps médian de survenue a été de 43 jours. Le patient a reçu un traitement endocrinien et arrêté le traitement par IMFINZI en raison du diabète sucré de type 1 immuno-médié. Une résolution de l'événement est survenue chez ce patient.
Hypophysite/ hypopituitarisme immno-médié(e)
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une hypophysite/ un hypopituitarisme immuno-médié(e) est survenu(e) chez 2 (< 0,1 %) patients, tous deux de grade 3. Le temps médian de survenue des événements était de 44 jours et de 50 jours. Les deux patients ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour) et un patient a interrompu le traitement par IMFINZI en raison de l'hypophysite/ hypopituitarisme immuno-médié(e).
Néphrite immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une néphrite immuno- médiée est survenue chez 9 (0,3 %) patients, y compris de grade 3 chez 2 (< 0,1 %) patients. Le temps médian de survenue a été de 87 jours (de 29 à 393 jours). Six (0,2 %) patients ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour) et 1 patient a également reçu du mycophénolate. Le traitement par IMFINZI a été interrompu chez 5 patients. La résolution est survenue chez 6 patients.
Réaction cutanée immuno-médiée
Dans la base de données combinées de tolérance d'IMFINZI en monothérapie, une éruption cutanée ou une dermatite immuno-médiée (incluant pemphigoïde) est survenue chez 45 (1,5 %) patients, y compris de grade 3 chez 12 (0,4 %) patients. Le temps médian de survenue a été de 41 jours (de 4 à 333 jours). Vingt patients sur 45 ont reçu un traitement par corticoïdes à haute dose (au moins 40 mg de prednisone ou équivalent par jour). Le traitement par IMFINZI a été interrompu chez 3 patients. La résolution est survenue chez 31 patients.
Réactions liées à la perfusion
Dans la base de données combinées de tolérance sur IMFINZI en monothérapie, les réactions liées à la perfusion sont survenues chez 49 (1,6 %) patients, y compris de grade 3 chez 5 (0,2 %) patients. Aucun événement de grade 4 ou 5 n'est survenu.
Immunogénicité
L'immunogénicité d'IMFINZI en monothérapie repose sur les données regroupées obtenues chez 2 280 patients traités par IMFINZI 10 mg/kg toutes les 2 semaines ou 20 mg/kg toutes les 4 semaines en monothérapie et chez qui la présence d'anticorps anti-médicament (anti-drug antibodies ou ADA) était évaluable. Soixante-neuf patients (3,0 %) ont eu un résultat positif au test des ADA apparus sous traitement. Les anticorps neutralisants (neutralizing antibodies ou nAb) dirigés contre le durvalumab ont été détectés chez 0,5 % (12/2 280) des patients. La présence des ADA n'a pas eu d'effet cliniquement pertinent sur la sécurité. Le nombre de patients est insuffisant pour déterminer l'impact des ADA sur l'efficacité. Sur la base de l'analyse de la population PK, une exposition légèrement inférieure chez les patients ADA positif est attendue toutefois, la diminution de l'exposition PK est inférieure à 30 % par rapport au patient type et n'est pas considérée comme cliniquement pertinente.
Dans l'étude CASPIAN, sur 201 patients traités par IMFINZI 1 500 mg toutes les 3 semaines en association avec une chimiothérapie et chez qui la présence d'ADA était évaluable, 0 (0 %) patient a eu un résultat positif au test des ADA apparus sous traitement. L'impact des ADA apparus sous traitement sur la PK, la sécurité clinique et l'efficacité du durvalumab n'a pas pu être évalué, car aucun échantillon patient n'a fourni de résultat positif au test des ADA du durvalumab apparus sous traitement.
Sujet âgé
Aucune différence globale de sécurité n'a été rapportée entre les patients âgés (≥ 65 ans) et les patients plus jeunes. Les données chez les patients atteints d'un CBPC-SE de 75 ans ou plus sont limitées.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet :signalement.social-sante.gouv.fr.
SURVEILLANCE du traitement :
- Tests de la fonction hépatique avant et périodiquement pendant le traitement.
- Tests de la fonction thyroïdienne avant et périodiquement pendant le traitement.
- Evaluation de la fonction rénale avant et périodiquement pendant le traitement.
CONTACTER IMMEDIATEMENT LE MEDECIN en cas de :
- Nouvelle toux, toux qui s'aggrave, essouflement, douleur dans la poitrine.
- Nausées, vomissements, baisse de l'appétit, douleur dans le côté
droit de l'estomac, jaunissement de la peau ou du blanc des yeux,
somnolence, urines foncées, saignements et bleus plus facile qu'en
temps normal.
- Diarrhée ou selles plus fréquentes, selles noirâtres, goudronneuses,
collantes ou contenant du sang ou des glaires, douleur ou sensibilité
sévère à l'estomac.
- Fréquence cardiaque rapide, fatigue extrême, prise ou perte de poids,
état vertigineux ou évanouissement, chute des cheveux, sensation de
froid, constipation, maux de tête persistants ou inhabituels.
- Sensation de faim ou de soif plus importante qu'en temps normal, miction plus fréquente.
- Diminution de la quantité d'urine.
- Eruption cutanée, démangeaisons, ampoules ou ulcères dans la bouche ou les autres muqueuses.
- Douleur dans la poitrine, essouflement ou battement cardiaque irrégulier.
- Douleurs ou faiblesse musculaires.
- Frissons ou tremblements, démangeaisons ou éruption cutanée, rougeur
de la peau, essouflement ou sifflement respiratoire, état vertigineux
ou fièvre.
CONTRACEPTION : les femmes susceptibles de procréer doivent utiliser
une méthode efficace de contraception pendant toute la durée du
traitement et la poursuivre pendant au moins 3 mois après la dernière
administration.
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines
(altération des capacités de concentration ou de réaction).
Femme en âge de procréer
Les femmes en âge de procréer doivent utiliser une méthode efficace de contraception pendant toute la durée du traitement par le durvalumab et la poursuivre pendant au moins 3 mois après la dernière administration de durvalumab.
Grossesse
Aucune donnée n'est disponible sur l'utilisation du durvalumab chez la femme enceinte. Sur la base de son mécanisme d'action, le durvalumab peut potentiellement impacter l'évolution de la grossesse. Dans un modèle murin allogénique de grossesse, la perturbation de la voie de signalisation PD-L1 a conduit à une augmentation des pertes foetales. Les études chez l'animal sur le durvalumab n'ont pas révélé de reprotoxicité (voir rubrique Données de sécurité précliniques). Il est connu que l'immunoglobuline humaine IgG1 traverse la barrière placentaire et le passage placentaire du durvalumab a été confirmé dans les études réalisées sur les animaux. L'administration du durvalumab chez la femme enceinte peut causer des dommages chez le foetus en conséquence son utilisation n'est recommandée ni pendant la grossesse ni chez les femmes en âge de procréer sans méthode efficace de contraception pendant le traitement et pour au moins 3 mois après la dernière dose.
Allaitement
On ignore si le durvalumab est sécrété dans le lait maternel. Les données toxicologiques disponibles chez les singes cynomolgus ont montré de faibles taux de durvalumab dans le lait maternel au 28ème jour après la naissance (voir rubrique Données de sécurité précliniques). Chez les humains, les anticorps peuvent être secrétés dans le lait maternel, toutefois le potentiel d'absorption et la nature des dommages éventuels chez le nouveau-né sont inconnus. Ainsi un risque potentiel pour l'enfant allaité ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement, soit d'interrompre ou de temporairement suspendre le durvalumab, en prenant en compte le bénéfice de l'allaitement pour l'enfant et le bénéfice du traitement pour la femme.
Fertilité
Aucune donnée n'est disponible sur les effets possibles du durvalumab sur la fertilité des humains ou des animaux.
L'utilisation de corticoïdes systémiques ou immunosupresseurs avant de débuter le durvalumab, à l'exception de dose physiologique de corticostéroïdes systémiques (≤ 10 mg/jour de prednisone ou équivalent), n'est pas recommandée en raison de son interférence possible avec l'activité pharmacodynamique et l'efficacité du durvalumab. Néanmoins, le traitement par corticoïdes systémiques ou immunosupresseurs peut être utilisé après avoir débuté le durvalumab pour traiter les effets indésirables liés à l'immunité (voir rubrique Mises en garde et précautions d'emploi).
Aucune étude pharmacocinétique (PK) d'interaction médicamenteuse formelle n'a été menée avec le durvalumab. Les voies d'élimination principales du durvalumab étant le catabolisme des protéines via le système réticuloendothélial ou de disposition médiée par la cible, aucune interaction médicamenteuse métabolique n'est attendue. Les interactions médicamenteuses PK entre le durvalumab et la chimiothérapie ont été évaluées dans l'étude CASPIAN et aucune interaction médicamenteuse PK cliniquement significative n'a été identifiée.
Le traitement doit être instauré et surveillé par un médecin ayant l'expérience du traitement des cancers.
Pendant la phase d'induction, IMFINZI est utilisé en association à la chimiothérapie (Etoposide+ sels de platine) toutes les 3 semaines pendant 4 cycles. La phase d'induction est suivie d'une phase d'entretien sans chimiothérapie au cours de laquelle IMFINZI est administré en monothérapie par perfusion intraveineuse toutes les 4 semaines.
Posologie
La dose recommandée d'IMFINZI selon la phase de traitement est présentée dans le Tableau 1. IMFINZI est administré sous forme de perfusion intraveineuse d'une durée d'1 heure.
Tableau 1. Dose recommandée d'IMFINZI
| Dose d'IMFINZI | Durée de traitement | |
| Phase d'induction | 1 500 mga en association avec une chimiothérapieb,c | toutes les 3 semaines (21 jours) pendant 4 cycles, |
| Phase d'entretien | 1 500 mga toutes les 4 semaines en monothérapie | Jusqu'à progression de la maladie ou toxicité inacceptable |
a Chez les patients pesant 30 kg ou moins, la dose doit être établie en fonction du poids, équivalant à 20 mg/kg d'IMFINZI en association avec une chimiothérapie toutes les 3 semaines (21 jours) pendant 4 cycles, suivis de 20 mg/kg toutes les 4 semaines en monothérapie jusqu'à ce que le poids soit supérieur à 30 kg.
b Administrer IMFINZI avant la chimiothérapie, le même jour.
c Lorsqu'IMFINZI est administré en association avec une chimiothérapie, se reporter aux informations de prescription concernant l'étoposide et le carboplatine ou le cisplatine, pour connaître la posologie appropriée.
Oubli ou retard de dose
Si au cours de la phase d'induction, une dose programmée d'IMFINZI et de chimiothérapie est oubliée ou retardée, l'association d'IMFINZI - chimiothérapie doit être administrée dès que possible. Si la chimiothérapie est administrée selon le calendrier prévu mais que la dose d'IMFINZI est oubliée ou retardée, il est alors recommandé d'attendre le prochain cycle planifié pour administrer la prochaine dose d'IMFINZI.
Si au cours de la phase de maintenance, une dose programmée d'IMFINZI est oubliée ou retardée, elle doit être administrée dès que possible. Il est recommandé de ne pas attendre la prochaine dose programmée. Le calendrier d'administration devra alors être modifié de manière à conserver un intervalle de 4 semaines entre les doses.
Modifications de dose pendant le traitement
Une augmentation ou une réduction de la dose n'est pas recommandée. La suspension de la dose ou l'arrêt du traitement peut être requis sur la base de la sécurité et la tolérance individuelles.
Les recommandations relatives au traitement des effets indésirables immuno-médiés sont décrites dans le tableau 2 (voir rubrique Mises en garde et précautions d'emploi).
Tableau 2. Modifications recommandées pour le traitement par IMFINZI et recommandations relatives au traitement
| Effets indésirables | Intensitéa | Modification du traitement par IMFINZI | Traitement par corticoïdes sauf indication contraire |
| Pneumopathie immuno- médiée / pneumopathie interstitielle | Grade 2 | Suspendre le traitement | Débuter le traitement par 1 à 2 mg/kg/jour de prednisone ou équivalent suivi d'une réduction progressive de la posologie |
| Grade 3 ou 4 | Interrompre définitivement | 1 à 4 mg/kg/jour de prednisone ou équivalent suivi d'une réduction progressive de la posologie | |
| Grade 2 avec | |||
| une ALAT ou une | |||
| ASAT | |||
| > 3-5 x LSN et/ou | |||
| une bilirubine | |||
| totale > 1,5- 3 x LSN | Suspendre le traitement | ||
| Grade 3 avec | |||
| une ASAT ou | |||
| une ALAT > 5- | Débuter le traitement par | ||
| ≤8 x LSN ou une | 1 à 2 mg/kg/jour de | ||
| Hépatite immuno-médiée | bilirubine totale > 3- ≤ 5 x LSN | prednisone ou équivalent suivi d'une réduction | |
| progressive de la posologie | |||
| Grade 3 avec une ASAT ou | |||
| une ALAT | |||
| > 8 x LSN ou une | |||
| bilirubine totale > 5 x LSN | Interrompre définitivement | ||
| ALAT ou ASAT | |||
| concomitante | |||
| > 3 x LSN et | |||
| bilirubine totale | |||
| > 2 x LSN sans |
| Effets indésirables | Intensitéa | Modification du traitement par IMFINZI | Traitement par corticoïdes sauf indication contraire |
| autre cause | |||
| Colite ou diarrhée immuno- médiée | Grade 2 | Suspendre le traitement | Débuter le traitement par 1 à 2 mg/kg/jour de prednisone ou équivalent suivi d'une réduction progressive de la posologie |
| Grade 3 ou 4 | Interrompre définitivement | ||
| Hyperthyroïdie immuno- médiée | Grade 2-4 | Suspendre le traitement jusqu'à l'obtention d'un état clinique stable | Traitement symptomatique, voir rubrique Effets indésirables |
| Hypothyroïdie immuno- médiée | Grade 2-4 | Pas de changements | Débuter une hormonothérapie thyroïdienne substitutive comme cliniquement indiqué |
| Insuffisance surrénalienne immuno-médiée ou hypophysite/hypopituitarisme | Grade 2-4 | Suspendre le traitement jusqu'à l'obtention d'un état clinique stable | Débuter un traitement par 1 à 2 mg/kg/jour de prednisone ou équivalent suivi d'une réduction progressive de la posologie et d'une hormonothérapie substitutive comme cliniquement indiqué |
| Diabète sucré de type 1 immuno-médié | Grade 2-4 | Pas de changements | Débuter le traitement par l'insuline comme cliniquement indiqué |
| Néphrite immuno-médiée | Grade 2 avec créatininémie > 1,5-3 x (LSN ou valeur initiale) | Suspendre le traitement | Débuter le traitement par 1 à 2 mg/kg/jour de prednisone ou équivalent suivi d'une réduction progressive de la posologie |
| Grade 3 avec une créatininémie > 3 x la valeur initiale ou > 3- 6 x LSN ; grade 4 avec une créatininémie > 6 x LSN | Interrompre définitivement | ||
| Éruption cutanée ou dermatite immuno-médiée (incluant pemphigoïde) | Grade 2 pendant > 1 semaine | Suspendre le traitement | Débuter le traitement par 1 à 2 mg/kg/jour de prednisone ou équivalent suivi d'une réduction progressive de la posologie |
| Grade 3 | |||
| Grade 4 | Interrompre définitivement | ||
| Myocardite immuno-médiée | Grade 2 | Suspendre le traitementb | Débuter le traitement par 2 à 4 mg/kg/jour de |
| Effets indésirables | Intensitéa | Modification du traitement par IMFINZI | Traitement par corticoïdes sauf indication contraire |
| Grade 3 ou 4, ou tout grade avec une biopsie positive | Interrompre définitivement | prednisone ou équivalent suivi d'une réduction progressive de la posologie | |
| Myosite/ Polymyosite immuno- médiée | Grade 2 ou 3 | Suspendre le traitement | Débuter le traitement par 1 à 4 mg/kg/jour de prednisone ou équivalent suivi d'une réduction progressive de la posologie |
| Grade 4 | Interrompre définitivementc | ||
| Réactions liées à la perfusion | Grade 1 ou 2 | Interrompre ou ralentir la vitesse de perfusion | Une prémédication est à envisager pour la prophylaxie de réactions ultérieures liées à la perfusion |
| Grade 3 ou 4 | Interrompre définitivement | ||
| Infection | Grade 3 ou 4 | Suspendre le traitement jusqu'à l'obtention d'un état clinique stable | |
| Autres effets indésirables immuno-médiés | Grade 3 | Suspendre le traitement | Envisager une dose initiale de 1 mg/kg/jour à 4 mg/kg/jour de prednisone ou équivalent suivi d'une réduction de la posologie |
| Grade 4 | Interrompre définitivementd |
a Critères communs de terminologie pour les événements indésirables, version 4.03. ALAT : alanine aminotransférase ; ASAT :
aspartate aminotransférase ; LSN : limite supérieure de la normale.
b En l'absence d'amélioration dans les 3 à 5 jours malgré la prise de corticoïdes, la mise sous thérapie additionnelle immunosuppressive doit être initiée immédiatement. Jusqu'à résolution (grade 0), une réduction de dose graduelle des corticoïdes doit être initiée et continuée pendant au moins 1 mois, après laquelle IMFINZI peut être repris sur la base du jugement clinique.
c Interrompre définitivement IMFINZI si les effets indésirables ne se résolvent pas en grade ≤ 1 dans les 30 jours ou s'il y a des signes d'insuffisance respiratoire.
d Pour la myasthénie grave, s'il y a des signes de faiblesse musculaire ou d'insuffisance respiratoire, IMFINZI doit être définitivement interrompu.
En ce qui concerne les effets indésirables immuno-médiés suspectés, une évaluation appropriée doit être menée afin de confirmer l'étiologie et d'exclure d'autres étiologies. L'augmentation de la dose de corticoïdes et/ou l'utilisation additionnelle d'immunosuppresseurs systémiques est à envisager s'il y a aggravation ou absence d'amélioration. Lorsqu'une amélioration avec un grade ≤ 1 est obtenue, une diminution des corticoïdes devra être initiée et continuée pendant au moins 1 mois. Après suspension, IMFINZI peut être repris dans les 12 semaines si les effets indésirables se sont améliorés jusqu'à un grade ≤ 1 et la dose de corticoïdes a été réduite à ≤ 10 mg de prednisone ou équivalent par jour. IMFINZI doit être définitivement arrêté pour les effets indésirables immuno-médiés récurrents de grade 3 ou 4 (sévères ou menaçant le pronostic vital).
Pour les effets indésirables non immuno-médiés, il faudra envisager de suspendre le traitement par IMFINZI pour les effets indésirables de grade 2 et 3 jusqu'à un retour à un grade ≤ 1 ou retour à la situation initiale. IMFINZI doit être arrêté pour les effets indésirables de grade 4 (à l'exception des anomalies de laboratoires de grade 4, pour lesquelles la décision d'arrêter le traitement doit être basé sur les signes / symptômes cliniques associés et le jugement clinique).
Populations particulières
Population pédiatrique
La sécurité et l'efficacité d'IMFINZI chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Sujet âgé
Aucune adaptation posologique n'est requise chez le sujet âgé (≥ 65 ans) (voir rubrique Propriétés pharmacodynamiques). Les données disponibles chez les patients âgés de 75 ans ou plus sont limitées.
Insuffisance rénale
Aucune adaptation posologique d'IMFINZI n'est recommandée pour les patients présentant une insuffisance rénale légère ou modérée. Chez les patients atteints d'une insuffisance rénale sévère, les données sont trop limitées pour tirer des conclusions sur cette population (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Les données disponibles chez les patients présentant une insuffisance hépatique modérée à sévère sont limitées. La voie hépatique étant peu impliquée dans l'élimination du durvalumab, aucune adaptation posologique d'IMFINZI n'est recommandée chez ces patients en raison de l'absence de différence d'exposition attendue (voir rubrique Propriétés pharmacocinétiques).
Mode d'administration
IMFINZI est destiné à une utilisation intraveineuse. Il doit être administré en perfusion intraveineuse pendant une durée d'1 heure (voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination).
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
Flacon non ouvert 3 ans
Solution diluée
En cas d'utilisation non immédiate, la stabilité physico-chimique d'IMFINZI en cours d'utilisation a été démontrée pendant une durée ne dépassant pas 24 heures à une température comprise entre 2°C et 8°C ou 12 heures à température ambiante ne dépassant pas 25°C à partir de la ponction du flacon jusqu'au début de l'administration.
Précautions particulières de conservation :A conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler.
A conserver dans l'emballage d'origine à l'abri de la lumière.
Pour les conditions de conservation du médicament après dilution, voir la rubrique Durée de conservation
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il n'y pas d'information sur le surdosage avec durvalumab. Dans le cas d'un surdosage, les patients doivent être étroitement surveillés pour tous signes ou symptômes d'effets indésirables, et un traitement symptomatique adapté doit être mis en place immédiatement.
Classe pharmacothérapeutique : agents antinéoplasiques, anticorps monoclonaux, code ATC : L01XC28.
Mécanisme d'action
L'expression de la protéine ligand-1 de mort cellulaire programmée (PD-L1) est une réponse immunitaire adaptative qui permet aux tumeurs d'échapper à la détection et à la destruction par le système immunitaire. L'expression de PD-L1 peut être induite par des signaux inflammatoires (l'interféron gamma par exemple) et PD-L1 peut être exprimée à la fois sur des cellules tumorales et sur des cellules immunitaires associées à la tumeur dans un microenvironnement tumoral. PD-L1 inhibe la fonction et l'activation des lymphocytes T par l'interaction avec PD-1 et CD80 (B7.1). En se liant à ses récepteurs, PD-L1 réduit l'activité et la prolifération des lymphocytes T cytotoxiques et la production de cytokines.
Le durvalumab est un anticorps monoclonal de type immunoglobuline G1 kappa (IgG1κ) entièrement humain, qui bloque de manière sélective les interactions entre PD-L1 et à la fois PD-1 et CD80 (B7.1). Le durvalumab n'induit pas de cytotoxicité à médiation cellulaire dépendante des anticorps (ADCC). Le blocage sélectif des interactions entre PD-L1/PD-1 et PD-L1/CD80 augmente les réponses immunitaires antitumorales et augmente l'activation des lymphocytes T.
Efficacité et sécurité clinique
CBPC - Étude CASPIAN
L'étude CASPIAN était conçue pour évaluer l'efficacité d'IMFINZI avec ou sans trémélimumab en association avec l'étoposide et soit le carboplatine, soit le cisplatine. CASPIAN était une étude multicentrique randomisée menée en ouvert chez 805 patients atteints d'un CBPC-SE naïfs de traitement avec un indice de performance OMS/ECOG de 0 ou 1, présentant les critères requis pour recevoir une chimiothérapie à base de platine en traitement de première ligne du CBPC, avec une espérance de vie ≥12 semaines, ayant des métastases cérébrales asymptomatiques ou traitées, au moins une lésion cible selon les critères RECIST 1.1 et une fonction organique et médullaire adéquate. L'étude n'incluait pas de patients ayant des antécédents de radiothérapie thoracique ; des antécédents de déficit immunitaire primitif actif ; des affections auto-immunes, y compris un syndrome paranéoplasique (SPN) ; des affections auto-immunes ou inflammatoires documentées actives ou antérieures ; ayant utilisé des immunosuppresseurs systémiques dans les 14 jours précédant la première dose de traitement, à l'exception d'une dose physiologique de corticoïdes systémiques ; ayant une tuberculose ou une hépatite B ou C ou une infection à VIH active ; ou patients recevant un vaccin atténué vivant au cours des 30 jours précédant ou suivant le début du traitement par IMFINZI.
La randomisation était stratifiée par traitement à base de platine prévu au cycle 1 (carboplatine ou cisplatine).
Les patients ont été randomisés selon un rapport 1 : 1 : 1 pour recevoir :
· Bras 1 : IMFINZI 1 500 mg + trémélimumab 75 mg + étoposide et soit carboplatine, soit cisplatine
· Bras 2 : IMFINZI 1 500 mg + étoposide et soit carboplatine, soit cisplatine
· Bras 3 : soit carboplatine (ASC 5 ou 6 mg/mL/min), soit cisplatine (75-80 mg/m2) au Jour 1 et étoposide (80-100 mg/m2) par voie intraveineuse aux Jours 1, 2 et 3 de chaque cycle de 21 jours entre 4 et 6 cycles.
Pour les patients randomisés dans les bras 1 et 2, l'étoposide et soit le carboplatine, soit le cisplatine, étaient limités à 4 cycles, toutes les 3 semaines après la randomisation. IMFINZI en monothérapie était poursuivi jusqu'à progression de la maladie ou toxicité inacceptable. L'administration d'IMFINZI en monothérapie était autorisée au-delà de la progression de la maladie si le patient était cliniquement stable et tirait un bénéfice clinique tel que déterminé par l'investigateur.
Les patients randomisés dans le bras 3 étaient autorisés à recevoir un total de 6 cycles d'étoposide et soit de carboplatine, soit de cisplatine. À l'issue de la chimiothérapie, une irradiation crânienne prophylactique (ICP) était autorisée uniquement dans le Bras 3, à la discrétion de l'investigateur.
Des évaluations tumorales ont été réalisées à la semaine 6 et à la semaine 12 à partir de la date de randomisation, puis toutes les 8 semaines jusqu'à progression objective confirmée de la maladie. Les évaluations de la survie avaient lieu tous les 2 mois après l'arrêt du traitement.
Les critères principaux d'évaluation de l'étude étaient la survie globale (SG) sous IMFINZI + chimiothérapie (bras 2) vs. chimiothérapie seule (bras 3) et IMFINZI + trémélimumab + chimiothérapie (bras 1) vs. chimiothérapie seule (bras 3). Le principal critère secondaire d'évaluation était la survie sans progression (SSP). Les autres critères secondaires d'évaluation étaient le taux de réponse objective (TRO), points de référence de la SG et de la SSP, ainsi que les résultats rapportés par les patients (RRP). La SSP et le TRO ont été évalués par l'investigateur conformément aux critères RECIST v1.1.
Lors d'une analyse intermédiaire planifiée, IMFINZI + chimiothérapie (bras 2) vs chimiothérapie (bras 3) étaient inclus dans les limites d'efficacité du critère principal de la SG. Les résultats sont résumés ci-dessous.
Les caractéristiques démographiques et initiales de la maladie étaient bien équilibrées entre les deux bras de l'étude (268 patients dans le bras 2 et 269 patients dans le bras 3). Les caractéristiques démographiques initiales de la population globale de l'étude étaient les suivantes : hommes (69,6 %), âge ≥ 65 ans (39,6 %), âge médian 63 ans (étendue : 28 à 82 ans), Caucasiens (83,8 %), Asiatiques (14,5 %), Noirs ou Afro-Américains (0,9 %), Autres (0,6 %), Non Hispaniques ou Latino-Américains (96,1%), fumeurs actifs ou anciens fumeurs (93,1 %), personnes n'ayant jamais fumé (6,9 %), Performance Status ECOG 0 (35,2 %), Performance Status ECOG 1 (64,8 %), Stade IV 90,3 %, 24,6 % des patients ont reçu du cisplatine et 74,1 % des patients ont reçu du carboplatine. Dans le bras 3, 56,8 % des patients ont reçu 6 cycles de chimiothérapie et 7,8 % des patients ont reçu une ICP.
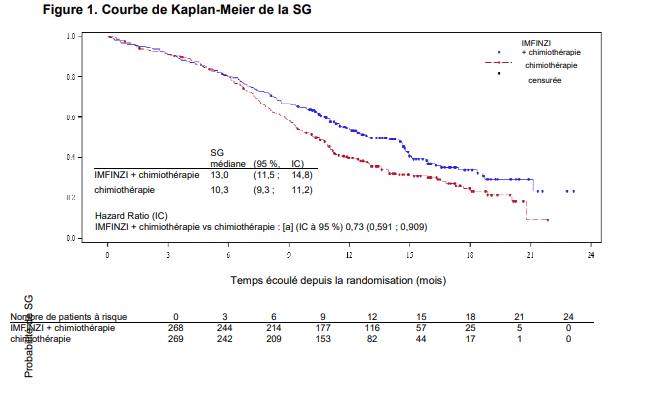
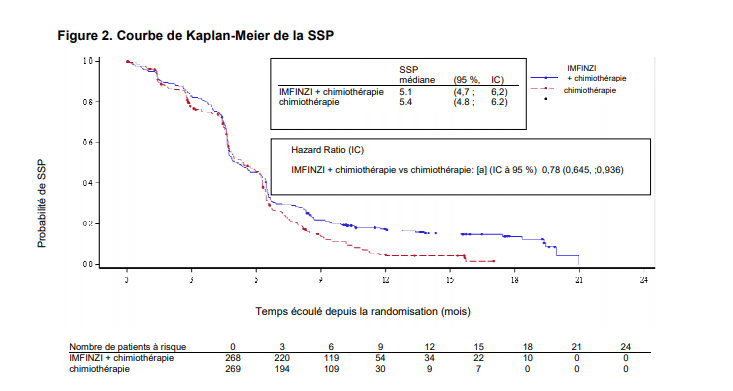
L'étude a démontré une amélioration statistiquement significative et cliniquement importante de la SG avec IMFINZI + chimiothérapie (bras 2) vs. chimiothérapie seule (bras 3) [HR = 0,73 (IC à 95 % : 0,591à 0,909), p = 0,0047]. IMFINZI + chimiothérapie a démontré une amélioration de la SSP vs. chimiothérapie seule [HR = 0,78 (IC à 95 % : 0,645 à 0,936) valeur p nominale =0,0078].
Voir Tableau 4 et Figures 1 et 2.
Tableau 4. Résultats d'efficacité de l'étude CASPIAN
| | Bras 2 : IMFINZI + étoposide et soit carboplatine, soit cisplatine (n = 268) | Bras 3 : étoposide et soit carboplatine, soit cisplatine (n = 269) | ||
| SG | | |||
| Nombre de décès (%) | 155 (57,8) | 181 (67,3) | ||
| SG médiane (mois) | 13,0 | 10,3 | ||
| (IC à 95 %) | (11,5 ; 14,8) | (9,3 ; 11,2) | ||
| HR (IC à 95 %)d | 0,73 (0,591, 0,909) | |||
| Valeur pc | 0,0047 | |||
| SG à 12 | mois | (%) | 53,7 | 39,8 |
| (IC à 95 %) | | | (47,4 ; 59,5) | (33,7 ; 45,8) |
| SG à 18 | mois | (%) | 33,9 | 24,7 |
| (IC à 95 %) | | | (26,9 ; 41,0) | (18,4 ; 31,6) |
| SSP | | |||
| Nombre d'événements (%) | 226 (84,3) | 233 (86,6) | ||
| SSP médiane (mois) | 5,1 | 5,4 | ||
| (IC à 95 %) | (4,7 ; 6,2) | (4,8 ; 6,2) | ||
| HR (IC à 95 %)d | 0,78 (0,645 ; 0,936) | |||
| Valeur pb | 0,0078 | |||
| SSP à 6 mois (%) | 45,4 | 45,6 | ||
| (IC à 95 %) | (39,3 ; 51,3) | (39,3 ; 51,7) | ||
| SSP à 12 mois (%) (IC à 95 %) | 17,5 (13,1 ; 22,5) | 4,7 (2,4 ; 8,0) |
| TRO n (%) (IC à 95 %)a | 182 (67,9) | 155 (57,6) |
| Réponse complète n (%) | 6 (2,2) | 2 (0,7) |
| Réponse partielle n (%) | 176 (65,7) | 153 (56,9) |
| Odds ratio (IC à 95 %)e | 1,56 (1,095 ; 2,218) | |
| Valeur pb | 0,0136 | |
| DdR médiane (mois) (IC à 95 %)a | 5,1 (4,9 ; 5,3) | 5,1 (4,8 ; 5,3) |
| DdR à 12 mois (%)a | 22,7 | 6,3 |
a Réponse objective confirmée.
b Valeur p nominale. La SSP était incluse dans la hiérarchie de la procédure de tests multiples (MTP) au second niveau. Elle n'a pas pu être testée au sein de la MTP car le bras 1 et le bras 2 devaient atteindre une signification statistique avant de passer à la SSP. Le TRO n'était pas inclus dans la MTP.
c Sur la base d'une fonction de dépense du risque alpha de Lan-DeMets avec une limite de type O'Brien Fleming avec le nombre réel d'événements observés, les limites pour la déclaration de la signification statistique sont de 0,0178 pour un alpha global de 4 % (Lan◦and◦DeMets 1983).
d L'analyse a été réalisée en utilisant le test du log-rank stratifié, avec ajustement en fonction du traitement à base de platine planifié lors du cycle 1 (carboplatine ou cisplatine), et en utilisant les tests de rank de l'approche d'association.
e L'analyse a été réalisée en utilisant un modèle de régression logistique avec ajustement en fonction du traitement à base de platine planifié lors du cycle 1 (carboplatine ou cisplatine) avec un IC à 95 % calculé par probabilité de profil.
Analyse en sous-groupe
Les améliorations de la SG en faveur des patients recevant IMFINZI + chimiothérapie comparativement à ceux recevant la chimiothérapie seule ont été constamment observées dans les sous-groupes prédéfinis analysés, y compris les caractéristiques démographiques, la région géographique, l'utilisation de carboplatine ou de cisplatine et les caractéristiques de la maladie.
Résultats rapportés par les patients
Les
symptômes, l'état fonctionnel et la qualité de vie liée à la santé
(HRQoL) rapportés par les patients ont été recueillis à l'aide de
l'échelle EORTC QLQ-C30 et de son module sur le cancer du poumon (EORTC
QLQ-LC13). Les deux questionnaires ont été évalués jusqu'à la seconde
progression de la maladie (SSP2) ou le décès (selon ce qui survenait en
premier). À l'inclusion, les symptômes rapportés par les patients,
l'état fonctionnel ou les scores HRQoL étaient comparables entre les
bras de l'étude. L'observance était de 60 % ou plus sur 84 semaines
dans le bras IMFINZI + chimiothérapie et 20 semaines dans le bras de
chimiothérapie seule.censurée
Prolongation du temps jusqu'à détérioration des symptômes, de l'état fonctionnel et de l'état de santégénéral/de la qualité de vie :
Le traitement par IMFINZI + chimiothérapie (bras 2) a démontré une amélioration en retardant la détérioration d'un large éventail de symptômes rapportés par les patients, de l'état fonctionnel et de l'état de santé général/de la QoL par rapport à la chimiothérapie seule (bras 3) (voir Tableaux 5 et 6). Le temps jusqu'à détérioration était défini comme le temps écoulé entre la date de randomisation et la date de la première détérioration cliniquement significative (aggravation ≥ 10 points par rapport à l'inclusion) confirmée lors d'une visite ultérieure ou le décès (quelle que soit la cause) en l'absence de détérioration cliniquement significative.
Tableau 5 : Prolongation du temps médian jusqu'à détérioration de l'état de santé général/de la QoL et de l'état fonctionnel (EORTC QLQ-C30)a
| | Prolongation du temps jusqu'à détérioration (mois) Bras 2 (N=261) vs. Bras 3 (N=260) |
| État de santé général/QoL | 8,4 vs, 7,2 0,81 (0,63 ; 1,05) ; p=0,1166 |
| Physique | 8,5 vs, 6,5 0,75 (0,58 ; 0,97) ; p=0,0276 |
| Cognitif | 8,4 vs, 6,0 0,61 (0,47 ; 0,78) ; p=0,0276 |
| Activité quotidienne | 7,4 vs, 5,9 0,71 (0,55 ; 0,90) ; p=0,0059 |
| Psychologique | 12,9 vs, 7,3 0,61 (0,46 ; 0,80) ; p=0,0003 |
| Social | 7,6 vs, 6,2 0,70 (0,55 ; 0,90) ; p=0,0048 |
a Valeur p du temps de détérioration basé sur le test du log-rank stratifié, sans ajustement pour multiplicité.
Tableau 6 : Prolongation du temps médian de détérioration des symptômes (EORTC QLQ-C30 et QLQ-LC13)a
| Échelle de symptômes | Prolongation du temps de détérioration (mois) Bras 2 (N=261) vs, Bras 3 (N=260) |
| Toux | 9,3 vs, 7,7 0,78 (0,60 ; 1,03) ; p=0,0747 |
| Dyspnée (QLQ-C30) | 9,0 vs, 7,4 0,75 (0,57 ; 0,99) ; p=0,0406 |
| Dyspnée (QLQ-LC13) | 6,5 vs, 5,5 0,79 (0,63 ; 1,01) ; p=0,0578 |
| Douleur | 7,8 vs, 6,7 0,79 (0,62 ; 1,02) ; p=0,0718 |
| Douleur thoracique | 10,6 vs, 7,8 0,76 (0,58 ; 1,00) ; p=0,0464 |
| Douleur au bras ou à l'épaule | 9,9 vs, 7,5 0,70 (0,54 ; 0,92) ; p=0,0088 |
| Douleur dans d'autres parties du corps | 7,8 vs, 6,4 0,72 (0,56 ; 0,92) ; p=0,0096 |
| Fatigue | 5,5 vs, 4,3 0,82 (0,65 ; 1,03) ; p=0,0835 |
| Insomnie | 8,6 vs, 7,3 0,75 (0,57 ; 0,98) ; p=0,0349 |
| Perte d'appétit | 8,3 vs, 6,6 0,70 (0,54 ; 0,90) ; p=0,0054 |
| Constipation | 11,1 vs, 7,3 0,65 (0,50 ; 0,86) ; p=0,0018 |
| Diarrhée | 14,6 vs, 7,7 0,59 (0,44 ; 0,77) ; p=0,0002 |
| Nausées/vomissements | 8,4 vs, 6,6 0,80 (0,63 ; 1,03) ; p=0,0809 |
| Hémoptysie | 18,3 vs, 10,5 0,64 (0,47 ; 0,88) ; p=0,0049 |
a Valeur p du temps de détérioration basé sur le test du log-rank stratifié, sans ajustement pour multiplicité.
Modification par rapport à l'inclusion des symptômes du cancer bronchique sur 12 mois (modèle mixteavec mesures répétées) :
IMFINZI + chimiothérapie a amélioré la perte d'appétit rapportée par le patient en démontrant une différence statistiquement significative de la modification moyenne par rapport à l'inclusion versus chimiothérapie seule pendant la période globale allant de la randomisation à 12 mois (différence moyenne estimée -4,5 ; IC à 99 % -9,04 ; -0,04 ; p=0,009). Les deux bras de traitement ont démontré une réduction numérique des symptômes en ce qui concerne la toux, la douleur thoracique, la dyspnée et la fatigue sur la même période.
Les données concernant les résultats rapportés par les patients doivent être interprétées en tenant compte de la méthodologie en ouvert de l'étude.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec le durvalumab dans tous les sous-groupes de la population pédiatrique dans le traitement des néoplasmes malins (à l'exception des tumeurs du système nerveux central, des néoplasmes du tissu lymphoïde et hématopoïétique) (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La pharmacocinétique (PK) du durvalumab a été évaluée à la fois pour IMFINZI en monothérapie et en association avec une chimiothérapie.
La PK du durvalumab administré par intraveineuse en monothérapie une fois toutes les deux, trois ou quatre semaines à des doses allant de 0,1 à 20 mg/kg, a été étudiée chez 2 903 patients porteurs de tumeurs solides. L'exposition PK a augmenté plus que proportionnellement à la dose (PK non linéaire) à des doses < 3 mg/kg et proportionnellement à la dose (PK linéaire) à des doses ≥ 3 mg/kg. L'état d'équilibre a été atteint à environ 16 semaines. D'après l'analyse de population PK qui a inclus 1878 patients ayant reçu le durvalumab en monothérapie à l'intervalle de dose ≥ 10 mg/kg toutes les 2 semaines, la moyenne géométrique du volume de distribution à l'état d'équilibre (Vss) était de 5,64 litres. La clairance du durvalumab (CL) a diminué avec le temps, ce qui s'est traduit par une moyenne géométrique de la clairance à l'état d'équilibre (CLss) de 8,16 ml/heure au jour 365 ; la diminution de la CLss n'a pas été considérée cliniquement pertinente. La demi-vie terminale (t1/2), d'après la CL initiale, était d'environ 18 jours. Il n'a pas été observé de différence cliniquement significative entre la PK du durvalumab administré en monothérapie et en association avec une chimiothérapie. Les voies d'élimination principales du durvalumab sont le catabolisme des protéines via le système réticuloendothélial ou de disposition médiée par la cible.
Populations particulières
L'âge (19 - 96 ans), le poids corporel (31 - 149 kg), le genre, le statut positif aux anticorps antimédicaments (ADA), les taux d'albumine, les taux de LDH, les taux de créatinine, la PD-L1 soluble, le type de tumeur, l'origine ethnique, ou le score de l'ECOG n'ont pas eu d'effet cliniquement significatif sur la pharmacocinétique du durvalumab.
Patients insuffisants rénaux
L'insuffisance rénale légère (clairance de la créatinine (CrCL) 60 à 89 ml/min) et l'insuffisance rénale modérée (clairance de la créatinine (CrCL) 30 à 59 ml/min) n'ont pas eu d'impact cliniquement significatif sur la PK du durvalumab. L'effet de l'insuffisance rénale sévère (CrCL 15 à 29 ml/min) sur la PK du durvalumab est inconnu.
Patients insuffisants hépatiques
L'insuffisance hépatique légère (bilirubine ≤ LSN et ASAT > LSN ou bilirubine >1,0 à 1,5 × LSN et n'importe quelle ASAT) n'a pas eu d'impact cliniquement significatif sur la PK du durvalumab. L'effet de l'insuffisance hépatique modérée (bilirubine >1,5 à 3 x LSN et n'importe quelle ASAT) ou de l'insuffisance hépatique sévère (bilirubine > 3,0 x LSN et n'importe quelle ASAT) sur la pharmacocinétique du durvalumab est inconnu toutefois, du fait que les anticorps monoclonaux IgG ne sont pas éliminés principalement par la voie hépatique, une modification de la fonction hépatique ne devrait pas influer sur l'exposition au durvalumab.
Le durvalumab n'a pas d'influence ou a une influence négligeable sur la capacité à conduire un véhicule et à utiliser des machines.
Carcinogénicité et mutagénicité
Le potentiel carcinogène et génotoxique du durvalumab n'a pas été évalué.
Toxicologie de la reproduction
Comme cela a été rapporté dans la littérature, la voie PD-1/PD-L1 joue un rôle central dans le maintien de la grossesse en conservant la tolérance immunitaire maternelle au foetus, et dans les modèles de gestation allogénique chez la souris, la perturbation de la voie de signalisation PD-L1 s'est traduite par une augmentation de la mort foetale. Dans les études de reproduction chez l'animal, l'administration du durvalumab à des singes cynomolgus femelles gestantes depuis la confirmation de la grossesse jusqu'à la mise bas, à des niveaux d'exposition environ 18 fois supérieurs à ceux observés à la dose clinique de 10 mg/kg de durvalumab (d'après l'AUC), a été associée à un transfert placentaire mais n'a pas été associée à une toxicité maternelle ni à des effets sur le développement embryofoetal, l'issue de la grossesse ou le développement post-natal. Des niveaux négligeables de durvalumab ont été trouvés dans le lait du singe cynomolgus le 28ème jour après la naissance.
Préparation de la solution
IMFINZI se présente sous la forme d'un flacon à usage unique et ne contient aucun conservateur, la technique d'asepsie doit être utilisée.
· Inspectez visuellement le médicament afin de déceler d'éventuelles particules ou une décoloration. IMFINZI est une solution claire à opalescente, incolore à jaune pâle. Jetez le flacon si vous remarquez que la solution est trouble, présente une décoloration ou contient des particules visibles. Ne pas secouer le flacon.
· Prélevez le volume requis du(des) flacon(s) d'IMFINZI et transférez-le dans une poche intraveineuse (IV) contenant du chlorure de sodium à 9 mg/ml (0,9 %), solution pour injection, ou du glucose 50 mg/ml (5 %), solution pour injection. Renverser délicatement afin de mélanger la solution diluée. La concentration finale de la solution diluée doit être comprise entre 1 mg/ml et 15 mg/ml. Ne pas congeler ou secouer la solution.
· Jeter toute solution inutilisée restant dans le flacon.
Administration
· Administrer la solution pour perfusion par voie intraveineuse pendant 1 heure à l'aide d'une ligne de perfusion intraveineuse contenant un filtre 0,2 ou 0,22 micron en ligne stérile à faible fixation protéinique.
· Ne pas administrer simultanément d'autres médicaments par la même ligne de perfusion. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament réservé à l’usage hospitalier.
Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant le traitement.
Solution à diluer pour perfusion (concentré stérile).
Solution claire à opalescente, incolore à jaune pâle, exempte de toute particule visible.
3 x 10 ml de solution à diluer dans 3 flacons en verre de type 1 muni d'une fermeture en élastomère et d'un opercule en aluminium blanc constituant un bouchon amovible contenant 3 x 500 mg de durvalumab. 1 boîte de 3 flacons.